
В России об орфанных заболеваниях заговорили относительно недавно. И больше всего обсуждают спинальную мышечную атрофию, тогда как сотни людей с другими, не менее серьезными заболеваниями также нуждаются в помощи и огромных средствах на лечение. «Такие дела» поговорили с людьми с редкими генетическими заболеваниями и узнали, как живут те, у кого нет надежды на выздоровление

18-летняя Даша Кошман хрупкая, как бабочка. Ее тонкие руки практически полностью спрятаны под одеждой, но на шее и лице видны свежие раны. У девушки редкое генетическое заболевание — буллезный эпидермолиз, при котором на теле и слизистых образуются пузыри и эрозии, а при любом механическом воздействии начинает слезать кожа.
Первые признаки заболевания — пузыри и нарывы — появились у Даши практически сразу после рождения. Но в 2003 году врачи думали, что это просто инфекция.
«Ставили капельницы, кололи антибиотики, успокоительное, но ничего про этот диагноз не рассказывали, — вспоминает мама Даши Елена. — Мне казалось, что сейчас полежим чуть-чуть, приедем домой и все пройдет. Никто не говорил, что будет так тяжело жить дальше. Сейчас хотя бы в интернете можно посмотреть, а тогда я понятия не имела, что нас ждет».
По словам Елены, осознание, что болезнь Даши неизлечима, пришло к ней только спустя несколько лет, когда они сделали генетический тест в Германии. До этого девочку пытались лечить и травами, и мазями, и уколами — все бесполезно.
 ДашаИллюстрация: Анна Иванцова для ТД
ДашаИллюстрация: Анна Иванцова для ТД
«С тех пор как Даша родилась, я не могу выйти на работу, потому что с дочкой нужно находиться рядом постоянно. Особенно в детстве все мы старались максимально оберегать ее, следить, чтобы она не травмировалась. Однажды не уследили и она упала на руки. Кожа с ладоней начала слезать, как перчатки. Это было очень страшно», — рассказывает Елена.
Сама Даша сравнивает боль, которую испытывает практически постоянно, с агонией.
«Раны болят так, что аж до костей пробирает. Если оценивать по шкале от одного до десяти, то чувствуется на все сто. Жжет, щиплет, дергает», — описывает девушка.
«Люди останавливаются и тычут в меня пальцем»
В 2020 году, только по официальным данным, в России насчитывалось 36 434 человека с редкими (орфанными) заболеваниями. Но так как система государственного учета охватывает лишь небольшую часть «редких» пациентов, в действительности их может быть больше. Главные проблемы, с которыми они сталкиваются в течение жизни, — сложность диагностирования и затраты на лечение.
Из-за низкой распространенности (в России не более десяти случаев заболевания на 100 тысяч населения) редкие болезни зачастую сложно диагностировать. У врачей, особенно в регионах, нет так называемой орфанной настороженности, из-за чего людям годами не могут поставить точный диагноз.
Кроме того, далеко не все редкие диагнозы входят в перечень 14 высокозатратных нозологий, лекарственное обеспечение которых происходит за счет средств федерального бюджета. В таком случае их финансирование будет зависеть от возможностей бюджета каждого конкретного региона. Учитывая, что 72 из 85 субъектов РФ являются дотационными, выделять огромные средства на лечение «редких» пациентов могут далеко не все.
Мама Даши, у которой диагностировали буллезный эпидермолиз, говорит, что сама научилась обрабатывать раны дочери, накладывать специальные салфетки и фиксирующие бинты. Но все эти препараты не входят в утвержденные перечни медицинских изделий, отпускаемых льготным категориям граждан за счет средств федерального бюджета. Поэтому деньги приходится искать самостоятельно.
 ДашаИллюстрация: Анна Иванцова для ТД
ДашаИллюстрация: Анна Иванцова для ТД
«Если не экономить и ухаживать как следует, то Даше требуется в месяц от 300 до 500 тысяч на мази и бинты, — отмечает мама. — В год — от трех до пяти миллионов. Собираем с миру по нитке: помогают нам фонд “Дети-бабочки” и родители детей с таким же диагнозом. Приходится экономить, отрезать маленькие кусочки, чтобы подольше хватило. В минздраве Приморского края мне сразу сказали, что таких денег нет».
Даше сложно выстраивать дружеские отношения и какие-либо социальные связи: многие боятся идти на контакт, потому что не понимают, что с ней. Даже в маленьком поселке, где все друг друга знают, она до сих пор сталкивается с неадекватной реакцией окружающих.
«Все раны перебинтованы и прикрыты одеждой, кроме лица, шеи и рук. Но всем обязательно нужно спросить: “А что с тобой? А это не заразно?” Иногда даже люди останавливаются на улице и тычут в меня пальцем, громко обсуждают», — замечает девушка и с грустью добавляет, что давно уже к этому привыкла.
Даша признается, что хотела бы жить в таком большом городе, как Москва, потому что «там просто всем все равно».
«Врачи говорили, что я до 18 лет не доживу»
День 23-летнего Дениса Гончарова расписан по минутам: встреча с представителями компании Google, переговоры с иностранными инвесторами, благотворительные проекты и интервью. Уже 20 лет Денис передвигается в инвалидном кресле, но ведет максимально насыщенную жизнь.
«Для меня как будто бы всегда так и было: я не помню, как ходил самостоятельно. Только фрагменты — то, как спотыкался, падал, передвигался по стеночке», — рассказывает молодой человек.
В три года ему диагностировали миодистрофию Дюшенна — генетическое заболевание, постепенно приводящее к дегенерации мышц. Параллельно у таких больных развиваются проблемы с сердцем и дыханием, из-за чего смерть в среднем наступает в возрасте 12—25 лет.
 ДенисИллюстрация: Анна Иванцова для ТД
ДенисИллюстрация: Анна Иванцова для ТД
«Меня даже из детского сада выгнали, потому что не хотели брать на себя никакую ответственность, — вспоминает молодой человек. — Поэтому маме пришлось уйти с работы, чтобы сидеть со мной. В семье работала только бабушка, и мы жили на очень маленькие деньги».
Денис окончил дистанционную школу и поступил в Юго-Западный государственный университет в Курске на программиста. Однажды у него произошел конфликт с одним из преподавателей, после чего Дениса обвинили в «поддержке оппозиции» — из-за того, что он был подписан на некоторые паблики во «ВКонтакте».
«Он [преподаватель] начал меня очень жестко гнобить, называть “врагом народа”. А потом на меня завели уголовное дело за клевету, — рассказывает Денис. — Я предал эту ситуацию огласке, потому что так не должно быть. Раздулось настолько, что даже вмешался губернатор. В итоге дело закрыли, а меня позвали учиться в ИТМО в Санкт-Петербурге».
Сегодня Денис параллельно совмещает несколько проектов: работает журналистом, контент-менеджером в СМИ. Но главная его цель — стартап NOLI Music — умная гитара, которая позволит играть начинающим музыкантам и людям с ограниченными возможностями здоровья. Компания зарегистрирована в США, сейчас идут переговоры с местными инвесторами.
Болезнь ограничивает молодого стартапера только физически: он не может покинуть квартиру без посторонней помощи. Денис понимает, что у людей с его диагнозом мышцы разрушаются, это необратимый процесс, но, по его собственным ощущениям, мышцы слабели до 16 лет, а потом как будто бы замерли в одном состоянии.
 ДенисИллюстрация: Анна Иванцова для ТД
ДенисИллюстрация: Анна Иванцова для ТД
Никакого лечения миодистрофии Дюшенна современная медицина не предлагает. Единственная помощь от государства — бесплатное инвалидное кресло. Но, по словам Дениса, это самый дешевый вариант, приводящий к еще большей травматизации — искривлению позвоночника. На пенсию по инвалидности в 13 тысяч, как он отмечает, «можно умереть, а жить нельзя».
«С самого детства врачи говорили, что я до 18 лет не доживу. Эти слова мне не забыть никогда. В детстве очень расстраивался и пугался из-за этого, а сейчас с юмором как-то вспоминаю. Но по этой причине очень не люблю праздновать свой день рождения и не считаю даты. Просто делаю то, что должен делать. Там уже как получится», — заключает Денис.
«Спорт для слепых больших денег не приносит»
Баясхалан Будаев каждое утро начинает с пробежки в парке возле дома. Этот маршрут он знает наизусть и может ориентироваться без посторонней помощи. Но в незнакомых местах начинаются проблемы — Баясхалан практически ничего не видит, и с каждым годом зрение становится все хуже.
«Когда мне было три года, родители заметили, что по вечерам я смотрю только на свет и не реагирую на них, — вспоминает молодой человек. — Обратились к офтальмологам, которые поставили диагноз “амавроз Лебера” — очень редкий вид пигментной дегенерации сетчатки».
 БаясхаланИллюстрация: Анна Иванцова для ТД
БаясхаланИллюстрация: Анна Иванцова для ТД
Зрение падало постепенно. В детстве Баясхалан вел вполне обычный образ жизни — посещал школу и секцию по вольной борьбе. Правда, от борьбы пришлось отказаться, так как постоянные падения и удары головой стали одними из причин прогрессирования болезни.
Чтобы не забрасывать спорт, Баясхалан занялся легкой атлетикой. «У нас в Бурятии нет условий для тренировок незрячих спортсменов. Один манеж на всю республику, где тренируют 500 человек — попробуй с ними потолкаться. К тому же манеж — это постоянные повороты, а значит, в моем случае — травмы. Поэтому я бегаю в парке возле дома», — рассказывает Баясхалан.
Впервые он принял участие в соревнованиях для незрячих спортсменов в 2013 году и сразу же взял серебро чемпионата России. Затем было еще несколько соревнований и золотые медали, после чего молодой человек попал в паралимпийскую сборную страны.
В последние три года зрение Баясхалана стало ухудшаться еще быстрее. Левый глаз практически совсем перестал видеть, правый различает только мутные очертания. Остановить прогрессирование болезни поможет генотерапевтический препарат «Лукстурна», его планируют зарегистрировать в России до конца года.
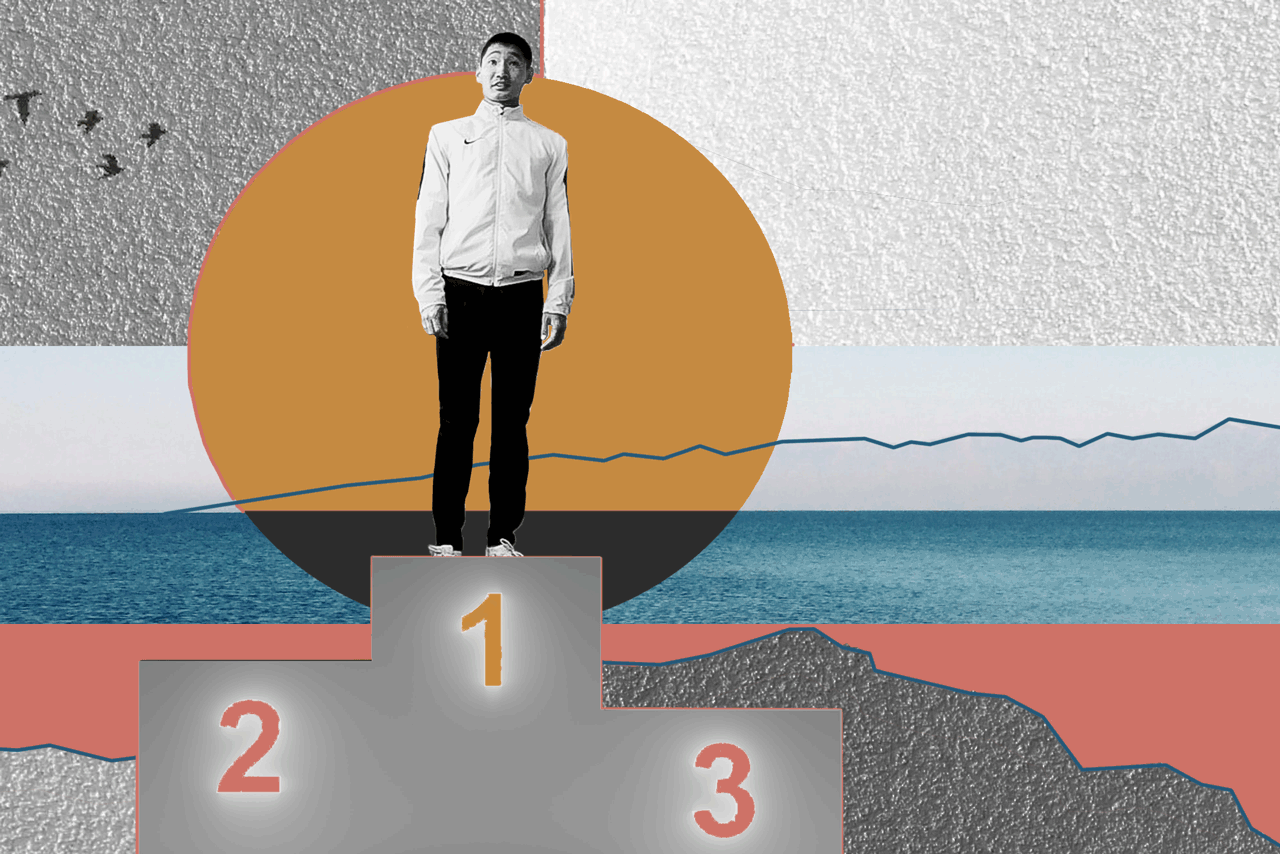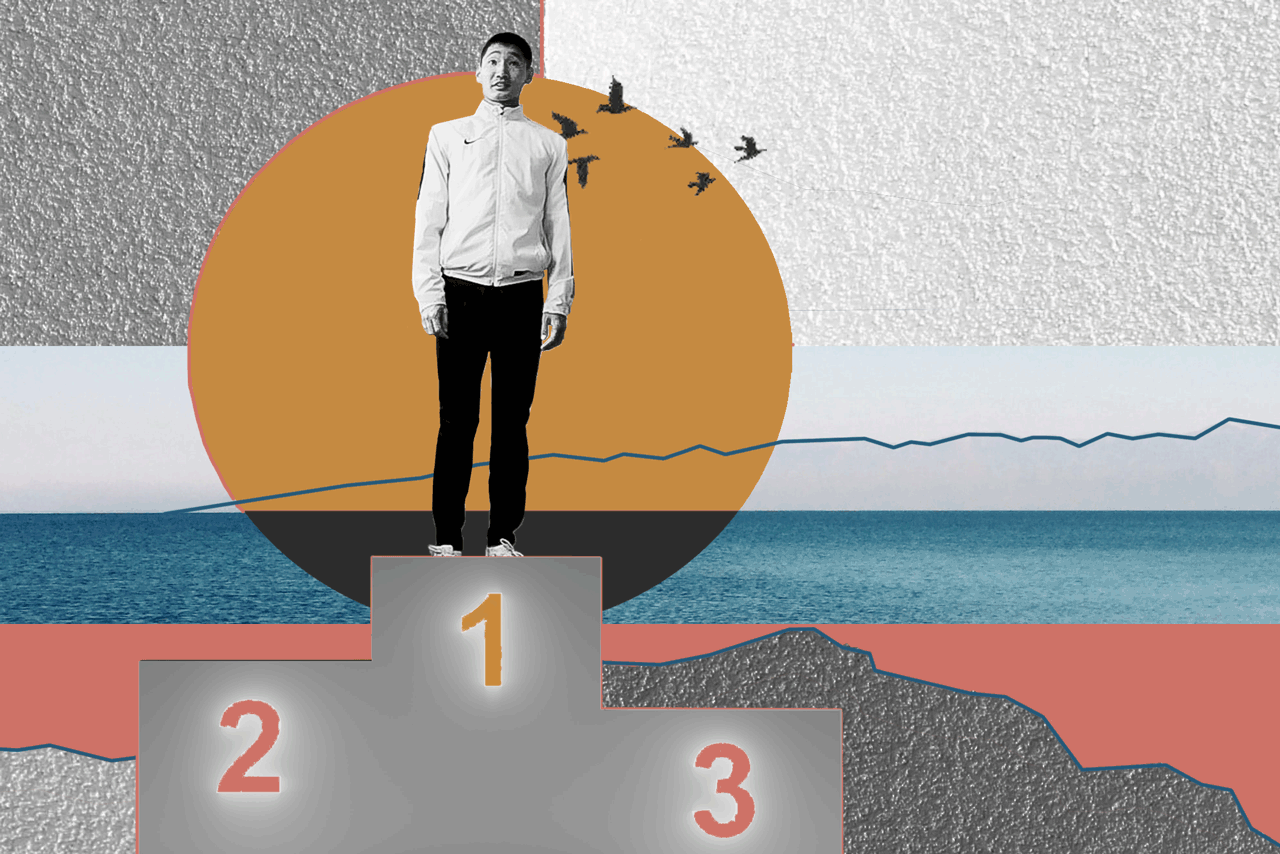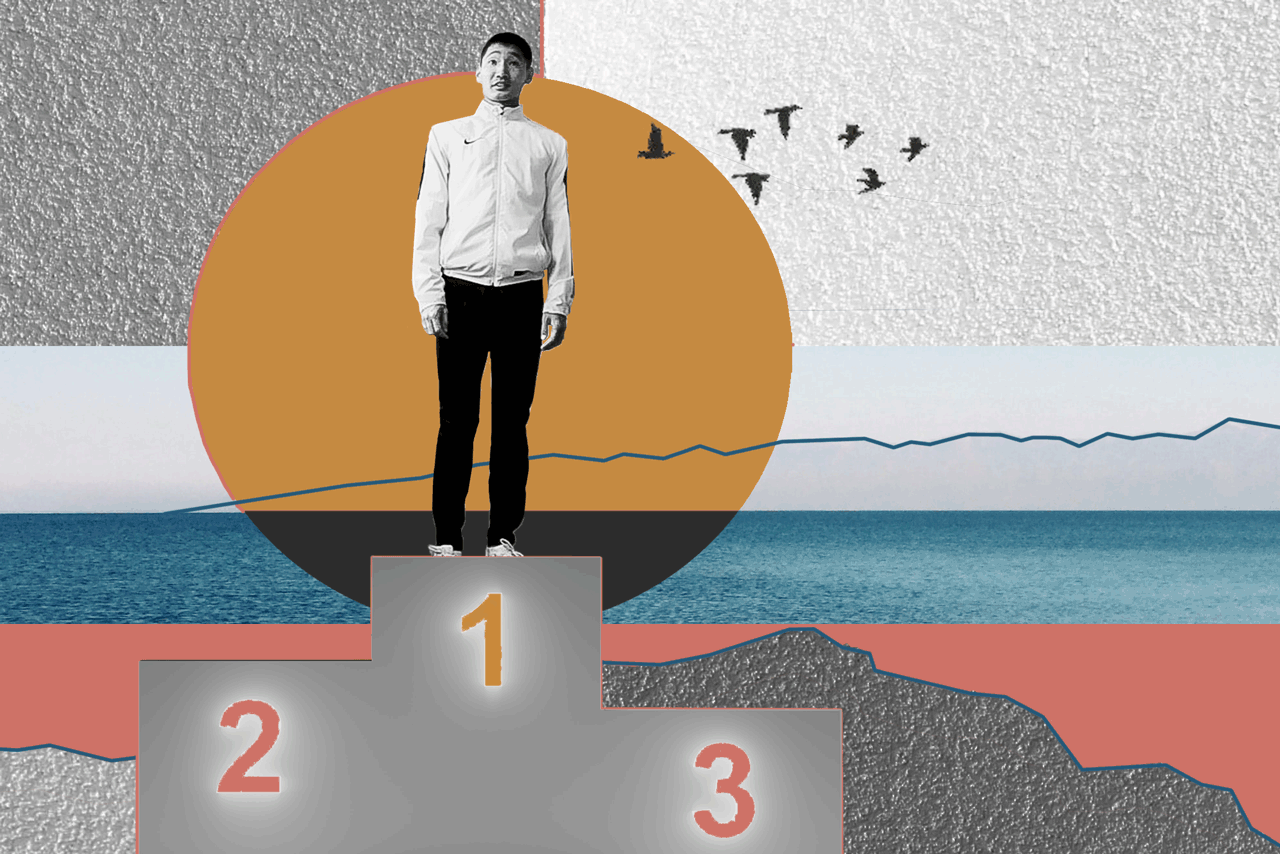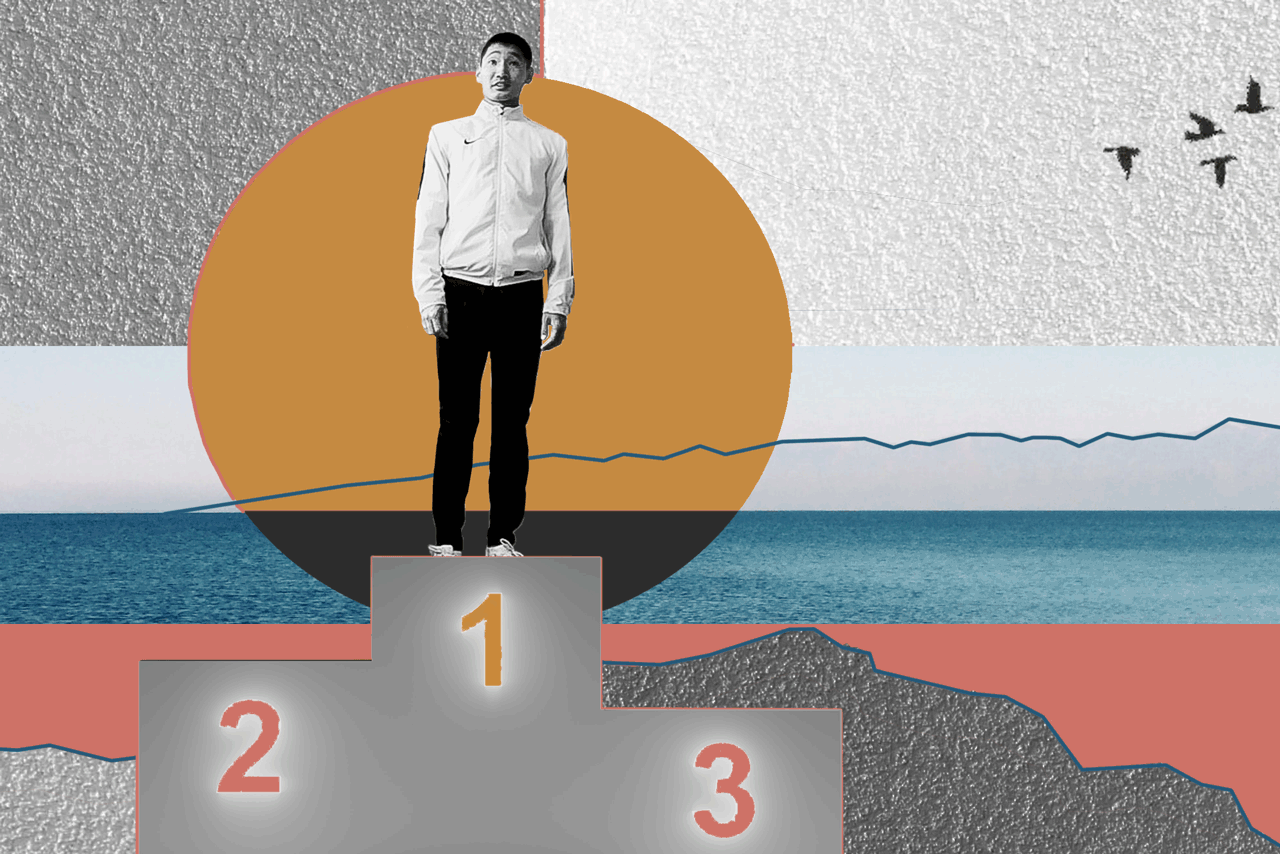 БаясхаланИллюстрация: Анна Иванцова для ТД
БаясхаланИллюстрация: Анна Иванцова для ТД
Один укол будет стоить около 25 миллионов рублей. Баясхалану необходимо сделать два. Так как амавроз Лебера не входит в перечень высокозатратных нозологий, на помощь Минздрава РФ и средства федерального бюджета рассчитывать не приходится.
«Если все получится, я буду первым взрослым человеком, кому сделают эту инъекцию в России, — замечает спортсмен. — Но как собрать столько денег? От государства я получаю только по две тысячи рублей в месяц. На работу не берут из-за инвалидности. Я сейчас пытаюсь развивать спорт для слепых в Бурятии, но больших денег это не приносит».
Текст создан в поддержку федеральной коммуникационной кампании «Редкое искусство помогать». Кампания носит просветительский характер и призвана рассказать обществу о трудностях лечения редких заболеваний, а также об особенных потребностях пациентов. «Такие дела» — информационный партнер кампании.
Еще больше важных новостей и хороших текстов от нас и наших коллег — в телеграм-канале «Таких дел». Подписывайтесь!
Каждый день мы пишем о самых важных проблемах в нашей стране и предлагаем способы их решения. За девять лет мы собрали 300 миллионов рублей в пользу проверенных благотворительных организаций.
«Такие дела» существуют благодаря пожертвованиям: с их помощью мы оплачиваем работу авторов, фотографов и редакторов, ездим в командировки и проводим исследования. Мы просим вас оформить пожертвование в поддержку проекта. Любая помощь, особенно если она регулярная, помогает нам работать.
Оформив регулярное пожертвование на сумму от 500 рублей, вы сможете присоединиться к «Таким друзьям» — сообществу близких по духу людей. Здесь вас ждут мастер-классы и воркшопы, общение с редакцией, обсуждение текстов и встречи с их героями.
Станьте частью перемен — оформите ежемесячное пожертвование. Спасибо, что вы с нами!
Помочь нам